
Note: The author would like to acknowledge the inestimable help received from conversations with Dr. John Lambshead, and for the enthusiastic support of Professor Eglund, whose analysis follows the article. There are probably several persons on the planet who will understand the analysis.
Many people have suggested that aging is a pre-programmed, genetically controlled function in higher animals. This appears to be confirmed by the research findings of Cynthia Kenyon, an eminently respectable scientist who publishes in peer-reviewed journals, on aging in nematodes. She reported extensions of nematode lifespan of five times normal, i.e. from around two weeks to ten, with their apparent vigour undiminished until shortly before death. In other words, they don't just drag out old age but function properly for an extended period.
She accomplishes this by selectively blocking the expression of various "DAF" genes, including DAF-2 and DAF-16. DAF-2 suppresses the action of DAF-16, the latter triggers or suppresses at least six other genes the end result of which is to promote longevity. DAF-16 seems to influence the production of proteins that protect against free radical damage. So it could be said that DAF-2 has the function of deliberately limiting a nematode's life span.
Of course, one has to be careful extrapolating from a nematode worm with a lifespan measured in weeks to something long lived like a human being. In nematodes, DAF-2 and DAF-16 are associated with moulting. Nematodes are Ecdysozoans (moulting animals) and these have the ability to go into a special 'shut down' state, known as 'dauer larvae' in the case of the Kenyon's test animal, the nematode Caenorhabditis elegans.
Human beings neither moult nor go into a shut down mode (not even teenagers). However, the DAF-2 gene is similar to a gene in mammals called IGF-1 that is connected with insulin function. Martin Holzenberger created strains of mice in which one or both copies of the rodent gene for the IGF-1 receptor had mutations. Mice lacking any normal copies died as embryos. However, mice with one working copy developed normally and lived, on average, 26 percent longer than did animals with two normal copies of the IGF-1 receptor gene.
For the purposes of this article, let us assume that getting old and dying is a defined process, a process that is controlled by our genes; that our life span is deliberately limited. Why? This seems astonishingly counter-evolutionary. The two basic driving forces in evolution are (i) survival and (ii) success in mating (for sexual organisms). Surviving by definition implies extension of life span. However, it is also true that for many organisms that the longer an organism survives the more successful it will be at mating – even geeks will manage to reproduce if given enough chances.
So if our life span is deliberately limited by our genes then there must be some evolutionary advantage. The rest of this article will try and address this point.
One possibility is that we limit our lifespan by the need for optimal performance up to the time of successful reproduction. This is the racing car analogy. An engineer designing a Ford or a Peugeot tends to over-engineer to give reliability and a decent lifespan rather than optimise for performance. In contrast, the racing genius Colin Chapman used to examine his cars after a race and any component that was in too good a condition was promptly lightened. An ideal Chapman racing car would fall to pieces one inch after crossing the finish line – but in first place!
The racing car analogy does not really seem to explain Kenyon's results because when DAF-2 is suppressed the worms stay younger longer; they do not stagger on in senility. It is almost as if the genes do not care how long a worm could live or what condition it might be in through most of that life but just decide it has lived long enough. What evolutionary mechanism could cause this?
In Utah, studies have been done that show that it is possible, in fact surprisingly easy, to track the presence of polygamous marriages by the occurrence of babies with birth defects. It turns out that having a few highly prolific males makes a surprisingly large impact on the occurrence of double recessives in the general population, and that this in turn leads to a surprisingly large number of birth-defects; children with Down's Syndrome, cleft palate, club feet, various leukodystrophies and the like occur much more commonly than they would without the prior presence of these males. This observation is anecdotally common human experience. Inbreeding causes an increase in nasty recessive genes in the population and polygamy inevitably will increase inbreeding.
This is a common problem in conservation. Once a population of an organism drops below a certain level then the species is in terrible trouble. In theory, one could recreate a species from a single male and female. In practice, there are likely to be enormous genetic issues.
For human populations, inbreeding is likely to be especially dangerous for two reasons. The first is that the human genome is particularly messy compared to other mammals such as dogs or rats. The second reason is that reproductive success of males in human beings (who are highly social animals) tends to be correlated with status and status tends to increase with age. As the hypothetical TV interviewer put to the young blonde, 'What first attracted you to this elderly, balding, multimillionaire, Miss Smith?' The wealthy middle-aged man with a younger trophy wife is a phenomenon observed in all human society. A long life span in men is more of an issue because one high status man can impregnate many women. Reason one will tend to accentuate the impact of reason two.
So if a meta-population (a subgroup) of a species that has mutations that shorten its lifespan has offspring that are more successful than the offspring of a longer-lived meta-population then the former population will replace the latter. An evolutionary mechanism, therefore, exists that could promote death genes.
Just because this is logical and reasonable does not make it true, but I leave you with one final thought. Women commonly live longer than men and, in my model, it is long lived men that should be more dangerous to the species. If you are male, then nature could have it in for you.
Classical and molecular genetics are concerned with the nature and transmission of genetic information, and how this information is translated into phenotypes. Population genetics theory is most successful when dealing with simply inherited traits - traits whose transmission follows simple Mendelian rules. Yet many of the most interesting and important traits are not so simply inherited: they depend on several genes, which often interact in complex ways with one another and with the environment.
Population genetics includes a large body of mathematical theory; one of the most richest and most successful bodies of mathematics in biology. The useful application of this theory has been greatly enhanced in recent years by new molecular techniques.
Usually, the first step is to study the frequency of different genotypes or phenotypes in a sample of the population. The concept locus is used to designate a chromosomal location, and the concept allele designates an alternative form of the gene occupying the considered position. Usually we are more interested in the frequencies of the different alleles than in the frequencies of the different genotypes. This is because allele frequency is a more economical way to characterise the population. The number of possible genotypes is enormous. Consider for example 100 loci, each with 4 segregating alleles. With 4 alleles A, B, C, D the total number of possible genotypes at each locus is 10: (1) four homozygotes: AA, BB, CC and DD plus (2) six heterozygotes: AB, AC, AD, BC, BD and CD. For all 100 loci there are thus 10100 possible genotypes, i.e. more than the number of atoms in our universe! It is, therefore, much better to stick to allele-frequencies.
The characterisation of a population in terms of allele frequencies rather than genotype frequencies has another advantage. In a Mendelian population, the genotypes are scrambled every generation by segregation and recombination. New combinations are put together only to be taken apart in later generations. If we are to study a population over a time period of more than a few generations, then the stable entity is the gene.
In nature it is the organism that survives and reproduces, so natural selection acts on the organisms and it is the fitness of the organism that determines the likelihood of survival and reproduction. But what an individual actually transmits to future generations is a random sample of its genes, and those genes that increase fitness will be more represented in future generations. Thus, there is selection of genes determining the properties like vigour and fertility.
Any description of nature, whether verbal or mathematical, will only be a caricature and therefore necessarily incomplete. In the previous century, scientists realised that we cannot ask whether a mathematical model is true, we can only ask if it gives a good or bad description of our data, and so may be used for prediction of future observations and experiments. Some models are only rough caricatures, but the advantage with this class of models is that they are easy to understand. An example is the successful theory of thermodynamics where the gas molecules are regarded as elastic spheres.
One of the most useful conventions in population genetics is the model of a gene pool. We assume that each parent contributes equally to a large pool of gametes. Each offspring is regarded as a random sample of one egg and one sperm from this pool. Each gene is chosen randomly, as if we were drawing different coloured beans from a bag. This oversimplified model building is called "beanbag genetics" and has a surprisingly strong predictive power. In 1964, the great British geneticist, J.B.S. Haldane, wrote an amusing and spirited article "A Defence of Beanbag Genetics" (Persp. Biol. & Med., 7: 343-359).
Clearly the beanbag model is a crude representation of nature. A real population has individuals of all ages, some dying, some choosing mates, some giving birth, etc, etc. Keeping track of all this information is not only impractical, but often is neither interesting nor important. Most questions of genetic or evolutionary interest do not require minute details. The gene pool model will give us the same kind of insight that simple models in the physical sciences do, and often with predictions that are sufficiently accurate for most uses. Further it is important to be aware of that departure from the simple models may usually be treated with appropriate modifications.
Suppose now that a population consist of individuals with an extremely long life span. In the tradition of "beanbag genetics" the population is homozygous at the locus determining life span; i.e. all genotypes are AA and code for long-life. However, in any population there will be some random death rate basically determined from outer sources like biological (virus, sickness, predation or accidents due, for example, to nature catastrophes). Thus, rather living forever, the long-lived strategy has to tolerate the operation of a random death rate; that is, the survival rate, S, is close to 1. This strategy is called evolutionary stable if the population rejects all kinds of invading alleles that reduce the life span. In order to investigate this stability, we assume that a small fraction %epsilon; of the gene pool consists of an allele 'a' such that the genotypes Aa induces a shorter life span via changes in the birth rates and survival rates. We must find the requirement that the new allele will increase in the gene pool.
Since we are only interested in the general principles that govern the stability of the long-life strategy we simplify the population dynamics of the mutant allele as follows: On average they live t years and reproduces bAa offspring of which lAa survives the next t years. The probability of surviving t years for the long-lived individuals is L = St = (S multiplied by itself t times). Finally, we ignore the homozygous mutant allele (of genotype aa), since their abundance is of the order %epsilon; * %epsilon; in the gene pool.
Thus the initial genotype frequencies are (1-%epsilon;) of AA and %epsilon; of Aa where the abundant first group of individuals (AA) have a very long life span and the second potential invader group (Aa) only live t years. In the gene pool the frequency of A is (1-%epsilon;)*1 + %epsilon;*(1/2) = 1-%epsilon;/2 while the frequency of a is ![]() .
.
We shall now try to answer the question: Under what circumstances is the fraction of the allele a expected to increase in the gene pool?
In order to solve this problem we need another assumption: The population size is so large that the mating may be considered as a random mixture of the genes. We must consider all kind of matings between the genotypes:
(1) Consider first a long-lived female (AA) that mates with a long-lived male (AA). During t years she produces bAA offspring (of genoype AA) that have a chance of lAA to survive the next t years. In the population the probability of such matings is (1-%epsilon;)*(1-%epsilon;).
(2) Consider next a long-lived female (AA) that mates with a mutant short-lived male (Aa). During t years she produces bAA offspring of which 50% are long-lived (AA) with a chance of lAA to survive the next t years, and 50% are short-lived (Aa) with a chance of lAa to survive the next t years . In the population the probability of such matings is (1-%epsilon;)*%epsilon;.
(3) Then consider a mutant short-lived female (Aa) that mates with a long-lived male (AA). During t years she produces bAa offspring of which 50% are long-lived (AA) with a chance of lAA to survive the next t years, and 50% are short-lived (Aa) with a chance of lAa to survive the next t years . In the population, the probability of such matings is %epsilon;*(1-%epsilon;).
(4) Finally consider a mutant short-lived female (Aa) that mates with a mutant short-lived male (Aa). During t years she produces bAa offspring of which 25% are long-lived (AA) with a chance of lAA to survive the next t years, and 50% are short-lived (Aa) with a chance of lAa to survive the next t years. In the population the probability of such matings is ??%epsilon;*%epsilon;.
After t years the number of long-lived and short-lived recruits are respectively
WAA = (1-%epsilon;)2 bAA lAA + (1-%epsilon;)*%epsilon; (1/2) bAA lAA + %epsilon;*(1-%epsilon;)(1/2) bAa lAA + ??%epsilon;*%epsilon;(1/4) bAa lAA
WAa = (1-%epsilon;)*%epsilon; (1/2) bAA lAa + %epsilon;*(1-%epsilon;)(1/2) bAa lAa + ??%epsilon;*%epsilon;(1/2) bAa lAa
Now, the number of adult long-lived individuals surviving a period of t years will be (1-%epsilon;)N *S where N is the total population size. In order to keep the population at a stable size of N individuals, we have to introduce a common death rate m for the recruits of all genotypes:
(1-m)*N*WAA + (1-m)*N*WAa + (1-%epsilon;)N *L = N
After t years the new fraction of the short-lived allele a in the gene pool is
![]()
Thus we see that the fraction of the allele a will increase in the gene pool if ![]() ,
,
i.e.
![]()
which is equivalent to
![]()
Reordering the terms, finally gives the requirement that the mutant allele coding for short-lived may invade the gene pool:
![]()
Thus, if the mutant allele a codes for a birth rate bAa and survival rate lAa such that this inequality is satisfied it will invade the gene pool. But what does this inequality essentially tell us? In order to see the essential requirement for invasion we may use standard techniques in numerical analysis. First, since %epsilon; is a small quantity in the order of 1/100, we may use the standard approximations in the final expressions: ![]()
so the inequality is practically the same as ![]()
Then we take a closer look at the extra non-specific mortality rate m that was introduced in order to keep the population at its equilibrium value N. In order to understand the magnitude of this quantity we consider the population without the mutant allele a (that is all genotypes are AA), so the equilibrium equation reduces to N*WAA + N *L = N which is seen to imply
Equilibrium condition:
WAA + L = 1
The equilibrium equation simply states that in order to stabilise the population, the recruitment to the stock must be balanced with the death rates. Now according to classical Darwinian thinking, successful new mutants only accomplish small changes. In our case, this means that the new allele a will only slightly modify the recruitment, so the final adjustment of the new short-lived recruits will be small. Hence
![]()
so we finally obtain the following essential criteria for successful invasion:
Criteria for successful invasion of the gene pool: ![]()
In words this criteria simply says that the new allele a will successfully invade the gene pool if the number of surviving mutant offspring to adulthood exceeds 1. Note that offspring of genotype Aa is created in two different ways: Either the mother is of genotype AA and mates with a male of genotype Aa in which case 50% of the offspring will be Aa, or the mother is of genotype Aa in which case also 50% of the offspring will be Aa. This explains why it is the average litter size of the two genotypes ![]() that enters the invasion criteria. Now the expected number of mutant offspring that reaches adulthood is found by multiplying with their survival rate lAa . Since the recruitment of the long-lived genotypes exactly matches the adult mortality 1 - L, the total recruitment to the adult group is exactly 1 (i.e. the equilibrium condition). Note that this is precisely where the long-life enters the equations: the total recruitment is the recruitment of offspring that manage to live up to adulthood PLUS the number of adults surviving the period (as mentioned above, only non-genetic accidents cause a small death rate among the long-lived individuals). So the criteria for successful invasion of the gene pool may be restated in words as follows:
that enters the invasion criteria. Now the expected number of mutant offspring that reaches adulthood is found by multiplying with their survival rate lAa . Since the recruitment of the long-lived genotypes exactly matches the adult mortality 1 - L, the total recruitment to the adult group is exactly 1 (i.e. the equilibrium condition). Note that this is precisely where the long-life enters the equations: the total recruitment is the recruitment of offspring that manage to live up to adulthood PLUS the number of adults surviving the period (as mentioned above, only non-genetic accidents cause a small death rate among the long-lived individuals). So the criteria for successful invasion of the gene pool may be restated in words as follows:
Criteria for successful invasion of the gene pool:
The recruitment of the short-lived individuals must exceed 1
Note that this is good classical Darwinism: The genotype with the highest recruitment will at the end fill up the whole gene pool.
It is now easy to see why Jim Baen's hypothesis is supported by this genetic model. The whole issue about why we must die may be formulated the question of whether a long-lived strategy is evolutionary stable. A simple, first approximation, genetic model says that the long-lived strategy will only be evolutionary stable if it is difficult for mutant alternatives to establish a recruitment larger than 1. But as Jim Baen has pointed on classical biological knowledge: there are all sorts of difficulties with inbreeding. Therefore almost any alternative strategy that reduces energy devoted to long-life and put it into clutch size or vitality of offspring will have a better total recruitment than the long-lived. Therefore, the strategy of eternal life is evolutionary unstable. For human beings this has the consequence that 125 years is the maximum age.
In this section we derive the same result in an alternative model that takes all the year-classes (cohorts) into account.
Suppose there are M adult long-lived individuals consisting of 1000 year classes, and let the clutch size per individual be bA , the survival up to maturity is lm and the survival rate of adults is sA . At equilibrium MbA offspring are produced each year by 1000 year classes:
![]()
which is seen to reduce to
Equilibrium condition
![]()
i.e. the population will be in equilibrium if the recruitment to the adult population (i.e. birth rate multiplied by immature survival) balances the adult death rate.
Consider now a potential invading allele a that codes for living only 100 years as adults, i.e. 10% of the prevailing strategy of 1000 years. Initially there are ![]() individuals of these new genotypes having a birth rate of ba , a survivorship of the immature period equal la and an adult survivorship of sa . With these parameters the iteration equation for the adult mutants are
individuals of these new genotypes having a birth rate of ba , a survivorship of the immature period equal la and an adult survivorship of sa . With these parameters the iteration equation for the adult mutants are
![]()
Substituting a general solution of the form ![]() gives the characteristic equation for the growth of the mutant
gives the characteristic equation for the growth of the mutant
![]()
In order to see where the root lies, define the two functions ![]()
and notice that
![]() ,
,
but since tm > tm-1 , f(r) will eventually become greater than g(r) . Thus somewhere they intercept, and the crucial question is whether they intercept to the left of r = 1 , in which case the number of invaders will decline, or to the right of r = 1 , in which case the number of short lived individuals will increase. Since
![]()
it follows that the condition for interception to the right of r = 1 , i.e. f(1) < g(1) ,is
![]() which finally gives
which finally gives
Criteria for successful invasion of the gene pool:
![]()
In words: The recruitment to the mutant short-lived individuals (=average birth rate of long- and short lived genotypes multiplied with the survival probability up to maturity) must be larger than the adult death rate of the short-lived individuals. Note that this condition is equivalent to that found in our previous Hardy-Weinberg type model, so the same discussion applies.
Let us finally see that if the following approximation of the population dynamics of the mutant
![]()
is substituted into the formula of the Hardy-Weinberg model, we arrive at the same criteria for %epsilon;M mutant to invade a homozygous population of (1-%epsilon;)M long-lived adults:
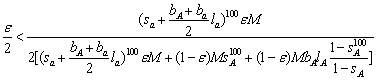
where the second term in the denominator is the number of long-lived adult that survived the total life span of the short lived (i.e. 100 years) and the third term is the number of adult long-lived that recruits to the adult population of long lived (since these are prevailing we ignore the contribution of long-lived from the short-lived). Using the equilibrium criterion, ![]() ,
,
we obtain
![]()
which is equivalent to ![]()
Thus, we arrive at precisely the same inequality, demonstrating an inner consistency in our models.
While the two previous models started with the assumption that long-life was an evolutionary stable strategy, we now develop a model where the different life history strategies are in direct competition with each other. However, if a new mutant code for better survival we shall now explicitly regard this as a strategy to allocate more energy to survival at the cost of energy devoted to reproduction. The simplest equation for such a trade-off model is
![]()
A first approximation to the population growth will then be
![]()
which has the solution
![]()
From this we see that natural selection will favour a strategy which maximises the function
![]()
Putting the derivative equal to zero,
![]() ,
,
gives the optimum survival rate as ![]()
implying an evolutionary stable expected life span of ![]()
So also in this model will the long-lived be selected against because they do not provide the optimal combination of birth rate and survival rate. Another consequence of tremendous importance is that given a strategy near the optimal, there will be no selection against deleterious genes at ages over T*. This allows death genes, racing-car analogy construction of organs, etc. Now human beings have evolved from species where these mechanisms already have been operating, so this is an evolutionary blind route with respect to long-life unless it is possible to enter our genes directly and reconstruct them. We are an animal that has evolved over many millions of years to be short lived.